Clustering Zheng et al 2017
This tutorial demonstrates how to replicate NCLUSION clustering results for ~94k pure peripheral blood mononuclear cells obtained from Zheng et al 2017 1. Open a bash terminal and follow along with the tutorial from your working directory.
Required Tutorial Installations
First, install NCLUSION into a Julia environment following the installation instructions here. The NCLUSION tutorials also require installation of additional R and python libraries. These packages are NOT required for running NCLUSION only.
The R environment
R is a software environment for statistical computing and graphics. The most recent version of R can be downloaded from the Comprehensive R Archive Network (CRAN). CRAN provides precompiled binary versions of R for Windows, macOS, and select Linux distributions. For detailed installation instructions for R and R libraries click here.
The following R libraries are required for replicating results in the NCLUSION tutorials:
- devtools
- reticulate
- ggplot2
- R.utils
- stringr
- Matrix
- Seurat
- dplyr
- RColorBrewer
- SingleCellExperiment
- tidyverse
- scales
- cowplot
- RCurl
- ComplexHeatmap
- circilize
- clusterProfiler
- enrichplot
- patchwork
The easiest method to install these packages is with the following example command entered in an R shell:
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(c("SingleCellExperiment", "ComplexHeatmap",
"clusterProfiler", "enrichplot"))
install.packages(c('patchwork',
'ggplot2',
'R.utils',
'stringr',
'Matrix',
'Seurat',
'dplyr',
'RColorBrewer',
'tidyverse',
'scales',
'cowplot',
'RCurl',
'circilize',
'reticulate',
'devtools'),
dependencies = TRUE);You can find a table of the R package versions here.
The Python Environment
We recommend that users install python packages into a new Anconda environment. Details for installing Anaconda and setting up a new environment can be found here.
Packages that are required to run NCLUSION tutorials include:
To install these packages, activate a new Anaconda environment and enter the following commands into a bash terminal:
conda install scikit-learn pandas matplotlib numpypython -m pip install scanpy anndata gseapy cvisudo apt-get install wgetAlternatively, you can download the environment with the exact versions by downloading this file: environment.yml and entering the following command into your bash terminal:
conda env create -f environment.ymlTutorial Scripts
Next, download the necessary tutorial scripts into your current working directory with the following bash commands. Alternatively, you can download the tutorial files as a zipped directory from here, transfer them to your working directory, and unzip to use.wget https://microsoft.github.io/nclusion/tutorial_scripts.zip
unzip tutorial_scripts.zipNOTE: to find more detailed documentation for each step of this tutorial, run each
bash script with the
--help flag.
Download and Preprocess Data
The Zheng + 2017 data contains scRNA-seq data for ~94k pure
peripheral blood mononuclear cells of 10 distinct cell types. The filtered, raw counts can be downloaded from here.
Alternatively, run the following wget commands to create a directory named
tutorial_data/zheng2017/ in your current working directory and download the filtered count files for each
cell type into that directory.
mkdir -p tutorial_data/
mkdir -p tutorial_data/zheng2017/
cd tutorial_data/zheng2017/
wget https://cf.10xgenomics.com/samples/cell-exp/1.1.0/cytotoxic_t/cytotoxic_t_filtered_gene_bc_matrices.tar.gz
wget https://cf.10xgenomics.com/samples/cell-exp/1.1.0/naive_cytotoxic/naive_cytotoxic_filtered_gene_bc_matrices.tar.gz
wget https://cf.10xgenomics.com/samples/cell-exp/1.1.0/cd56_nk/cd56_nk_filtered_gene_bc_matrices.tar.gz
wget https://cf.10xgenomics.com/samples/cell-exp/1.1.0/cd4_t_helper/cd4_t_helper_filtered_gene_bc_matrices.tar.gz
wget https://cf.10xgenomics.com/samples/cell-exp/1.1.0/memory_t/memory_t_filtered_gene_bc_matrices.tar.gz
wget https://cf.10xgenomics.com/samples/cell-exp/1.1.0/naive_t/naive_t_filtered_gene_bc_matrices.tar.gz
wget https://cf.10xgenomics.com/samples/cell-exp/1.1.0/regulatory_t/regulatory_t_filtered_gene_bc_matrices.tar.gz
wget https://cf.10xgenomics.com/samples/cell-exp/1.1.0/cd34/cd34_filtered_gene_bc_matrices.tar.gz
wget https://cf.10xgenomics.com/samples/cell-exp/1.1.0/b_cells/b_cells_filtered_gene_bc_matrices.tar.gz
wget https://cf.10xgenomics.com/samples/cell-exp/1.1.0/cd14_monocytes/cd14_monocytes_filtered_gene_bc_matrices.tar.gzThen, unzip the files in the data directory with the following bash command:
for f in `ls *.gz`; do
filename=$(basename -- "$f")
extension="${filename##*.}"
filename="${filename%.*}"
filename=$(basename -- "$filename")
filename="${filename%.*}"
echo "$filename";
mkdir "${filename}"
mv "${f}" "${filename}"
cd "${filename}"
tar -xzvf "$f"
cd ../
done
cd ../../ Before running NCLUSION to cluster the Zheng + 2017 cells, the data must be
converted to the AnnData type, and
concatenated into a single data object. Then, to preprocess the data, we filter out genes that were expressed in less than 3 cells, cells
with less than 200 genes expressed, greater than 20% mitochondrial reads, and
less than 5% ribosomal reads to obtain 94,615 high-quality cells representing 10
distinct cell types. We then subset the data to the top 5000 highly-variable
genes. The data conversion, concatenation, and preprocessing can be
executed by entering the following command into a bash terminal, which then
saves the
filtered, preprocessed data as an AnnData file located at
tutorial_data/zheng2017/5000hvgs_zheng2017_preprocessed.h5ad.
. tutorial_scripts/preprocess.sh --datapath="tutorial_data/zheng2017/" --savepath="tutorial_data/zheng2017/5000hvgs_zheng2017_preprocessed.h5ad" --data_name="zheng" --n_hvgs=5000Run NCLUSION
To run NCLUSION from your working direcorty, enter the following bash command.
NOTE: set the --julia_env variable to the
relative path to your Julia project environment where NCLUSION is installed.
mkdir -p zheng2017_outputs
. tutorial_scripts/run_nclusion.sh --datapath='tutorial_data/zheng2017/5000hvgs_zheng2017_preprocessed.h5ad' --julia_env='PATH/TO/JULIA/ENV' --data_name='zheng2017' --output_dir='zheng2017_outputs/'All tutorial output files can be found in the specified output
directory:
zheng2017_outputs/. All output files generated by NCLUSION can be
found in:
zheng2017_outputs/outputs/EXPERIMENT_nclusion_zheng2017/DATASET_zheng2017_5000HVGs-94615N/YEAR_MONTH_DATE_TIME/
NOTE: 'YEAR-MONTH-DATE-TIME' labels in the output path/files corresponds to the time stamp of when the results were generated. The time stamp is added automatically by NCLUSION.
In the output directory, you can find the following output files:
- zheng2017_5000HVGs-94615N_nclusion-YEAR-MONTH-DATE-TIME.csv
- .csv file that contains the NCLUSION clustering assignments, where each row displays relevant metadata and clustering results for each cell.
- The 'condition' column displays the cell's experimental condition labels (all cells in this study have the same condition label).
- The 'cell_id' column displays the cell's unique identifier label.
- The 'cell_type' column displays the cell's manual cell-type annotation published in this study. This column would be empty for unannotated datasets.
- The 'called_label' column displays the numerical equivalent of the 'cell_type' label, mapped automatically by NCLUSION.
- The 'inferred_label' column displays the cell's NCLUSION cluster assignment.
- 5000G-YEAR-MONTH-DATE-TIME-pips.csv
- .csv file that contains the Posterior Inclusion Probabilities (PIPs) of genes across clusters identified by NCLUSION. PIPs are used as a summary of evidence for a gene being associated with driving the identity of any phenotypic cluster. For a particular cluster, a higher PIP indicates that a gene is more significant to the formation of that cluster.
- _QuickSummary_YEAR-MONTH-DATE-TIME.txt
- .txt file that contains the input parameters that were used to run NCLUSION.
- output.jld2
- .jld2 file that contains all model parameters
- YEAR-MONTH-DATE-TIME-Nk.csv
- .csv file that contains estimated number of cells in each cluster identified by NCLUSION
NOTE: In the following analyses, clusters are named according to the cluster mapping file:
tutorial_scripts/cluster_mapping/zheng_mapping.csv which maps
the NCLUSION labels to labels in [1:number of clusters], for aesthetic
purposes. This mapping retains NCLUSION cluster memberships.
A. Quantitative Evaluation Metrics
To evaluate the NCLUSION clustering results, the following command calculates
various evaluation metrics: Jaccard Indexs, Adjusted Rand Index (ARI) (extrinsic), McIntosh Evenness, and Gini-Simpson Index (intrinsic).
Extrinsic metric calculations are dependent on
experimental annotations of the data, that act as the "true" labels, while intrinsic metrics do not. Therefore, extrinsic metrics are
only calculated when "true" labels are provided in the "cell_type" observation
layer of the input AnnData object. The metric calculations are
automatically saved to .csv files in the specified output
directory. Each file is labeled according to the following format: '{name_of_metric}_NCLUSION_pbmc_5000hvgs.csv'
To calculate evaluation metrics, enter the following command into your bash terminal.
NOTE: be sure to replace --input_path variable with the relative path to your NCLUSION clustering results and --project with the path to your Julia environment where NCLUSION is installed.
python tutorial_scripts/utility_scripts/make_intermediates.py --input_path "/PATH/TO/NCLUSION/CLUSTERING/FILE" --title "NCLUSION Zheng et al 2017" --output_path_file "zheng2017_outputs/" --output_path_figure "zheng2017_outputs/" --output_suffix '' --dataset_ordering_mode "pbmc" --col_sum_threshold '0.15'
julia --project='PATH/TO/JULIA/ENV' tutorial_scripts/utility_scripts/get_Julia_jaccard_metrics.jl "zheng2017_outputs/pbmc_NCLUSION_mapped_clusters.csv" '4' '6' '1' 'NCLUSION' '5000' "pbmc" "" 'jaccardtrans' "zheng2017_outputs/"
python tutorial_scripts/utility_scripts/make_evalmetrics.py --labels_path "zheng2017_outputs/pbmc_NCLUSION_mapped_clusters.csv" --data_path 'tutorial_data/zheng2017/5000hvgs_zheng2017_preprocessed.h5ad' --eval_metric "ginisimpsonindex" --condition_idx '1' --data_partition '' --dataset "pbmc" --method 'NCLUSION' --hvgs '5000' --output_path_file "zheng2017_outputs/" --output_suffix ''
python tutorial_scripts/utility_scripts/make_evalmetrics.py --labels_path "zheng2017_outputs/pbmc_NCLUSION_mapped_clusters.csv" --data_path 'tutorial_data/zheng2017/5000hvgs_zheng2017_preprocessed.h5ad' --eval_metric "mcintosheveness" --condition_idx '1' --data_partition '' --dataset "pbmc" --method 'NCLUSION' --hvgs '5000' --output_path_file "zheng2017_outputs/" --output_suffix ''
B. Generate Cell Distribution Heatmap
Next, generate a heatmap that displays how cells from each cell-type (determined by the "true" labels obtained from Zheng et al 2017) are distributed across clusters that are identified by NCLUSION. Each row, i, represents one cell-type, and each column, j, represents one cluster. Therefore the color intensity at location (i, j) on the heatmap corresponds to percent of cells with "true" cell-type label i that were assigned to cluster j by NCLUSION.
To generate the heatmap from the NCLUSION result, enter the following commands into a bash terminal.
NOTE: be sure to replace --nclusion_results input with the relative path to your NCLUSION clustering results.
NOTE: if you want to load packages from a specific R library, append --rlib="/PATH/TO/R/LIBRARY" onto
this command.
. tutorial_scripts/make_heatmap.sh --nclusion_results="/PATH/TO/NCLUSION/CLUSTERING/FILE" --pct_table="zheng2017_outputs/cell_distribution_table.csv" --cluster_dict="tutorial_scripts/cluster_mapping/zheng_mapping.csv" --save_heatmap="zheng2017_outputs/cell_distribution_heatmap.pdf" --data_name="zheng"This will produce a pdf file named zheng2017_outputs/cell_distriubtion_heatmap.pdfcontaining the cell distriubtion heatmap shown
below. The
column labels do not correspond to NCLUSION's cluster labels. The script labels
each cluster with their positional values in the heatmap for aesthetic purposes. A
mapping of the original NCLUSION cluster labels to these new positional labels can
be found in a file named tutorial_scripts/cluster_mapping/zheng_mapping.csv

C. Annotated TSNEs
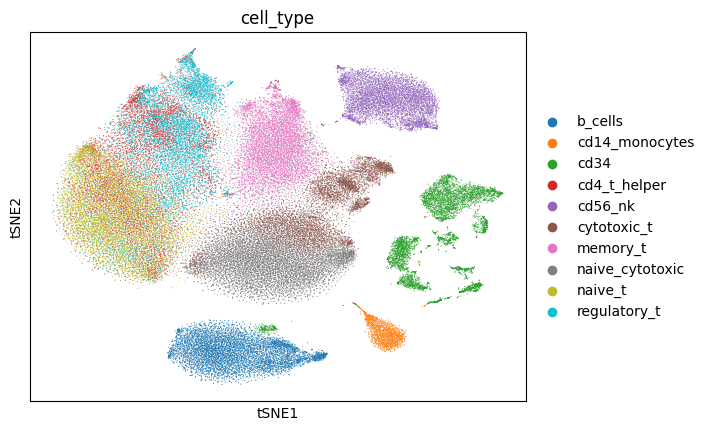
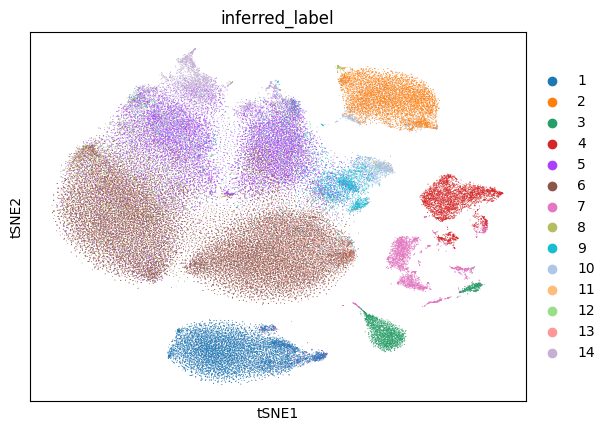
Now, we will plot the t-distributed stochastic neighbor embedding (tSNE) for the data annotated with various labels: the called cell type labels that were published along with the data, and the NCLUSION-inferred cell type labels. We will also generate tSNEs corresponding to the boolean label of each cells' membership to a given NCLUSION-inferred cluster, such that cells assigned to cluster n are colored and all other cells are greyed out. The total number of tSNEs produced are equal to the total number of NCLUSION-inferred clusters plus two. Run the following command to generate these plots.
NOTE: be sure to replace --nclusion_results input with the relative path to your NCLUSION clustering results.
. tutorial_scripts/make_tsnes.sh --datapath="tutorial_data/zheng2017/5000hvgs_zheng2017_preprocessed.h5ad" --nclusion_results="/PATH/TO/NCLUSION/CLUSTERING/FILE" --data_name="zheng2017"
mv figures/ zheng2017_outputs/All tsnes produces by this script can be found in the output directory:
zheng2017_outputs/figures/. The tSNEs colored by called
cell-type, inferred cell-type, and Cluster 1 membership are shown below.

D. Variable Selection Analysis
Next, analyze results of NCLUSION's variable selection functionality to gain insight into the biological mechanisms driving the formation of each cluster. The reulting plots are described in detail in the following sub-sections. To generate these plots, enter the following command into a bash terminal.
NOTE: if you want to load packages from a specific R library, append --rlib="/PATH/TO/R/LIBRARY" onto
this command.
NOTE: be sure to replace --nclusion_results, --pips, and --path_to_nk inputs with the relative path to your clustering assignment, PIP, and Nk results files (generated by NCLUSION).
. tutorial_scripts/analyze_results.sh --datapath="tutorial_data/zheng2017/5000hvgs_zheng2017_preprocessed.h5ad" --pips="/PATH/TO/PIPS/FILE" --nclusion_results="/PATH/TO/NCLUSION/CLUSTERING/FILE" --output_dir="zheng2017_outputs/" --data_name='zheng' --mapping="tutorial_scripts/cluster_mapping/zheng_mapping.csv" --path_to_nk="PATH/TO/NK/FILE"All plots produced by this script can be found in the specified output
directory: zheng2017_outputs/
directory. Each plot is described in detail below.
i. Adjusted Posterior Inclusion Probability (PIP) Heatmap
We generated a heatmap that displays adjusted Posterior Inclusion
Probabilities (PIPs) of genes across
clusters identified by NCLUSION. We use PIPs as a summary of evidence for gene being associated with driving the identity of any phenotypic cluster.
For a particular cluster, a
higher PIP indicates that a gene is more significant to the formation of that
cluster. Before generating this heatmap, the plotting script adjusted the PIP
values to account for
the number of clusters in which it is deemed significant. For example, if a gene
is deemed to be significant in driving all clusters, its PIP score will be
scaled down, as it is less helpful for identifying important differences between each
cluster compared to unique significant genes that are identified in fewer clusters. Only
genes with adjusted PIPs above a threshold of 0.5 are included in this analysis.
The plot is saved to zheng2017_outputs/heatmap_AdjustedPips_KbyG_defaultclustering.pdf.

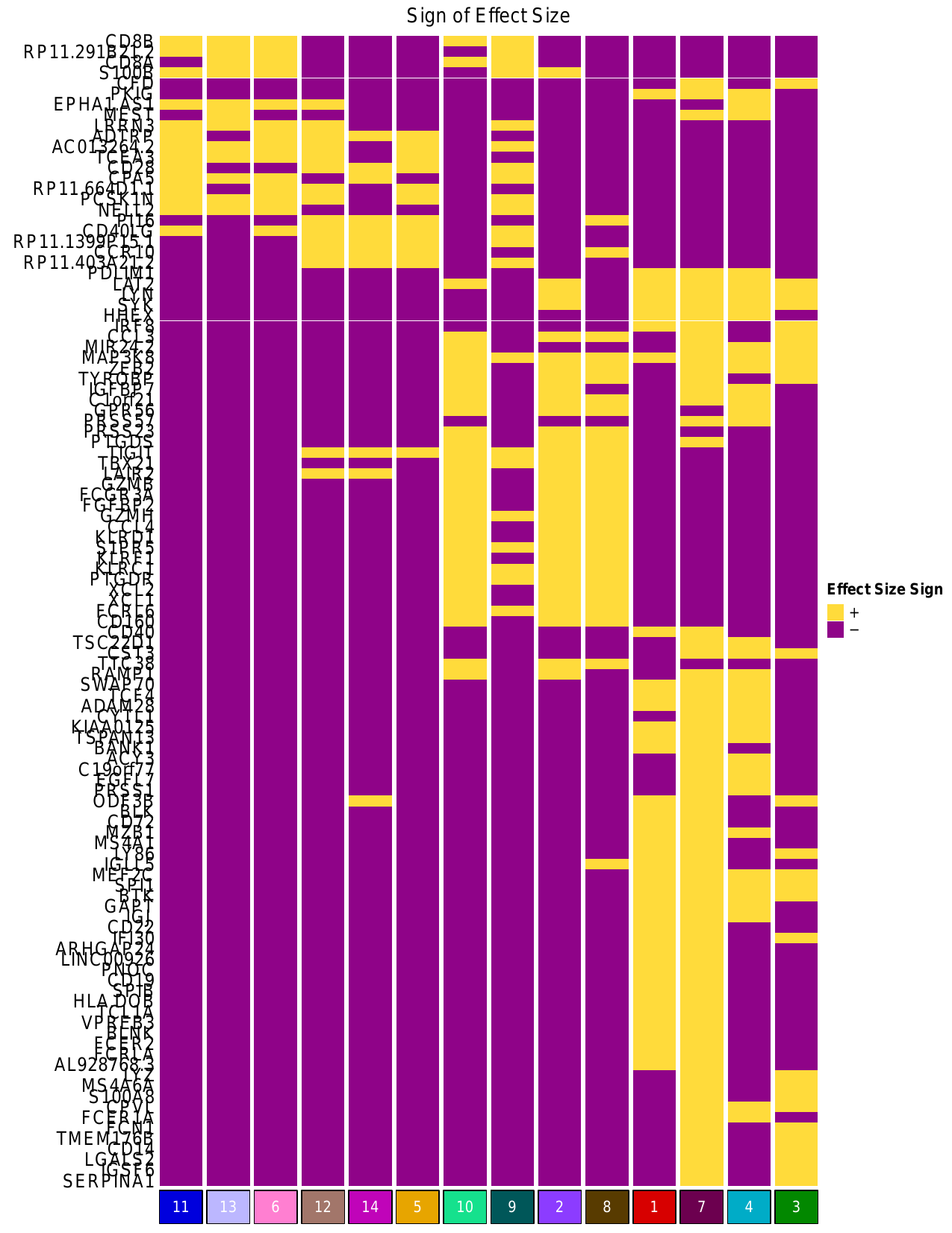
ii. Effect Size Heatmap
The script also generated heatmap representing the effect size of the genes included
in the adjusted PIP heatmap, which is shown below. For a given cluster, the effect size
indicates the direction of expression (+ or -) for each gene in that cluster.
The plot is saved to zheng2017_outputs/heatmap_EffectSizeSignMat_KbyG_basedonAdjPips.pdf
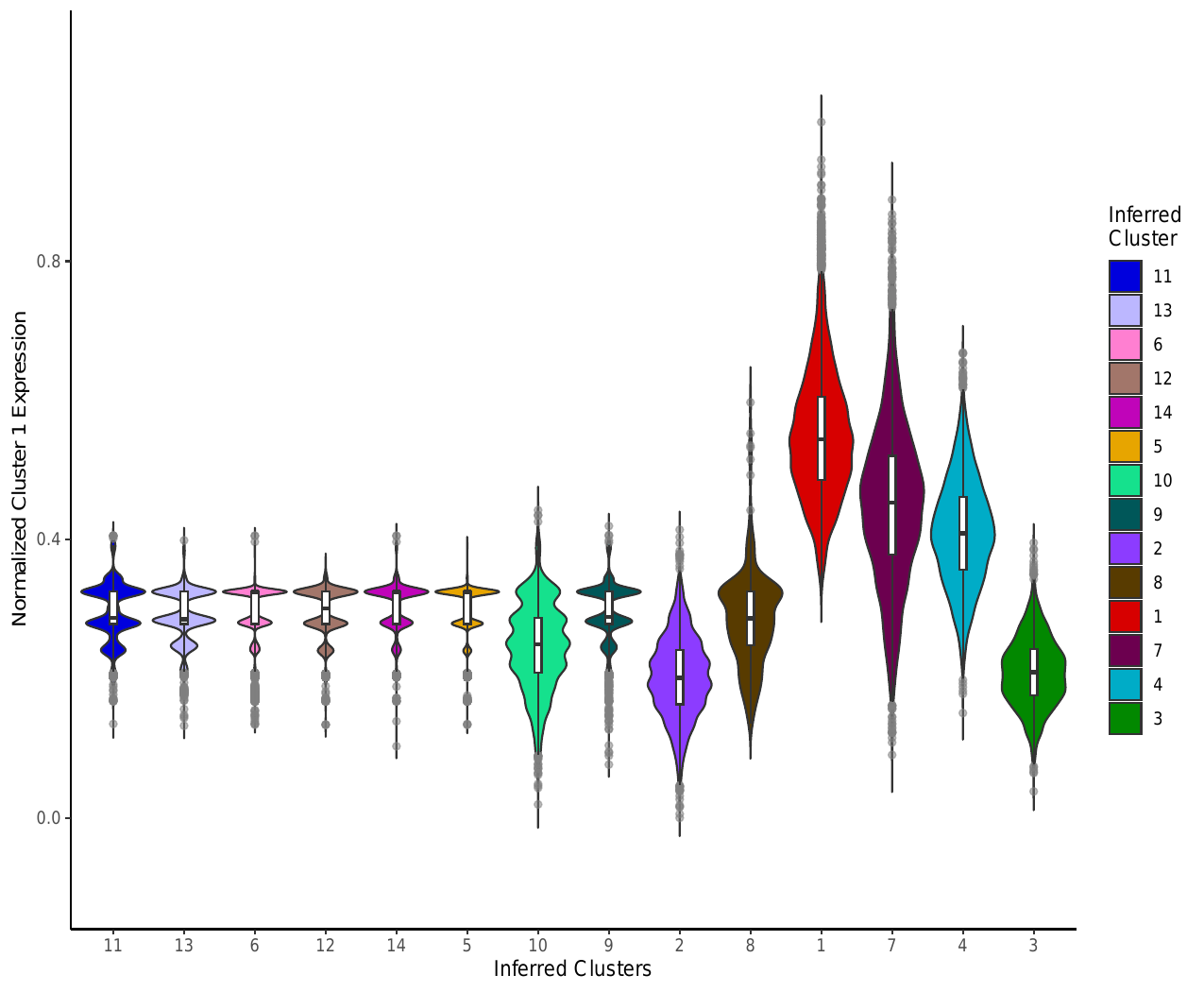
iii. Module Expression Violin Analysis
In addition to the heatmaps, this script generated module expression violin
plots for each cluster. Each violin plot compares the expression of
significant genes identified in a particular cluster to the expression of
that group of genes in the other clusters. The module expression analysis for
Cluster 1 is shown below. The plots are saved to
zheng2017_outputs/violinplot_tsne_normedmodulescore_colored_nolegend_Cluster_NGenes.pdf
where N is equal to the cluster label.
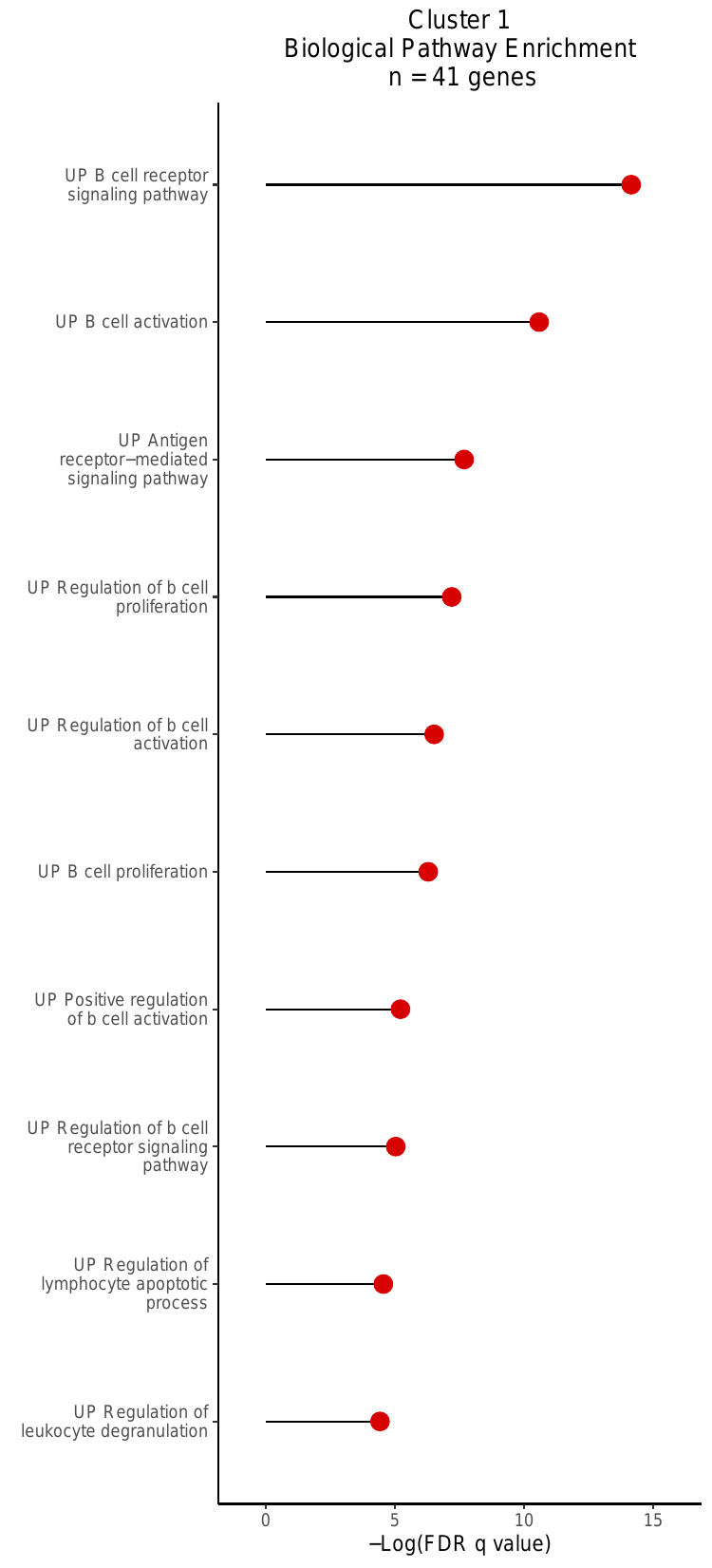
v. Gene-set Over-Enrichment (GO) Analysis
Lastly, this script produced biological pathway
enrichment plots. For each cluster, this analysis uses a
hypergeometric test to calculate the enrichment of genes in a supplied gene
set (genes with significant PIPs [>0.5] in that cluster). The GO analysis
for Cluster 1 is shown below. The plots are saved to
zheng2017_outputs/GO_bp_enrichmentplots_Cluster_NGenes.pdf
where N is equal to the cluster label.
References
Grace X. Y. Zheng, Jessica M. Terry, Phillip Belgrader, Paul Ryvkin, Zachary W. Bent, Ryan Wilson, Solongo B. Ziraldo, Tobias D. Wheeler, Geoff P. McDermott, Junjie Zhu, Mark T. Gregory, Joe Shuga, Luz Montesclaros, Jason G. Underwood, Donald A. Masquelier, Stefanie Y. Nishimura, Michael Schnall-Levin, Paul W. Wyatt, Christopher M. Hindson, Rajiv Bharadwaj, Alexander Wong, Kevin D. Ness, Lan W. Beppu, H. Joachim Deeg, Christopher McFarland, Keith R. Loeb, William J. Valente, Nolan G. Ericson, Emily A. Stevens, Jerald P. Radich, Tarjei S. Mikkelsen, Benjamin J. Hindson, and Jason H. Bielas. Massively parallel digital transcriptional profiling of single cells. Nature Communications, 8(11):14049, Jan 2017. ISSN 2041-1723. doi: 10.1038/ ncomms14049 https://doi.org/10.1038/ncomms14049↩︎